

Abstract
1. Introduction
DNA microarray technology can provide a broad picture of the state of the cell, by monitoring the expression level of thousands of genes at the same time [1]–[2]. Analysis of these new data requires mathematical tools that are adaptable to the large quanties of data, while reducing the complexity of the data to make them comprehensible. So far, some signal processing approaches such as singular value decomposition (SVD/PCA) and independent component analysis (ICA) have been proposed to analyze genome ― wide expression data [3]–[5]. However, these two methods require pre-processing of the micro-array data. Although normalization and transformation can reduce noise in micro-array experiments, it is possible for more noise and artifacts to appear in the data as a result of pre-processing. One of NMF advantages is that it can directly process these data without normalization, which make the analysis results more confident. Here we propose that the NMF approach is employed to analyze micro-array expression data. The uses of NMF in gene expression pattern extraction are explored. The design and implement of the optimal NMF algorithm are described. To verify the efficiency of the approach, NMF is employed to analyze
- a set of synthetic micro-array data with known characteristic patterns and
- two biological datasets that have cell cycle-regulated genes.
2. Method
The equation for non - negative matrix factorization of VV is the following: V=W\cdot H \eqno{\hbox{(1)}}V=W⋅H(1)
Where WW is of dimension mm by rr and HH is of dimension rr by nn. The desired rank rr is chosen so that (m+n) < mn(m+n)<mn, and the product WHWH can be regarded as a compressed form of the data in VV. In practical terms, this factorization finds a small set of “basis vectors” WW and a set of “hidden” describing factors HH. Since the columns of HH are in one ― to ― one correspondence with the columns of VV, the results can be interpreted as each column of VV being described as weighted of a few basis vectors, the weights being the corresponding column of HH. Our analysis shows that the vector in the lth row of the matrix H, {e_{l}}H,el lists the expression of the lth eigengene across the different arrays. So, we infer that an eigengene {e_{l}}el represent a characteristic transcription response pattern across all arrays, while this pattern is biological interpretable. The vector in the lth column of the matrix W, {a_{l}}W,al lists the genome ― wide expression in the lth eigenassay. By analogy we infer that the eigenassay {a_{l}}al represents the cellular state that corresponds to this pattern, and this cellular state have certain biological meaning. Thus, the characteristic expression patterns that are biologically meaningful can be directly extracted from the transformation matrices HH based on the above inference.
We design an optimal algorithm for NMF approach. Compared with the existing NMF algorithms, our algorithm is more stable and converges very fast. We have coded the final algorithm in highly optimized C.
3. Results
Some simulation experiments are made in order to verify the efficiency of NMF algorithm.
In simulations, NMF is employed to analyze a set of synthetic microarray data with
known characteristic patterns and two biological datasets that have been processed
by SVD approach and are thus thought to be gold standards. Here the results for the
\alpha-α− factor dataset with 6108 genes are given as an example. The characteristic patterns
extracted from the \alpha-α− factor dataset are shown in Fig. 1, where eigengene 1 fits an exponent function, primarily resembling a steady decay,
eigengene 2 shows an increasing sharply and decreasing slowly expression, superimposed
on expression oscillation during the cell cycle, eigengene 3 and 4 fits non ― normalized
sine and cosine function of period T\approx 67T≈67 min respectively. We infer they represent cell cycle ― regulated transcript patterns.
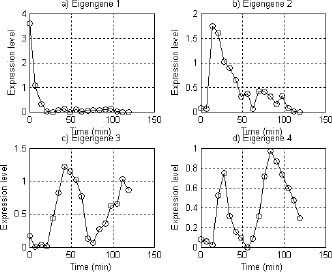
Fig. 1.Fig. 1.
4. Conclusions
The simulation experiments show that NMF performs very well for the above datasets and is robustness under high noisy level. We found that, like SVD, an important ability distinguishing NMF and related methods from other analysis methods is the capability to detect weak signals in the data. Even when the structure of the data does not allow separation of data points, causing clustering algorithms to fail, NMF can extract biologically meaningful patterns. Obviously, these interesting expression patterns are helpful to investigate the related biologically problems further.
Finally, our conclusions are that NMF can be used as a powerful tool to extract the biologically meaningful expression patterns from genomic ― wide expression data.
Reference
- [1]R. J. Cho, M. J. Campbell, “A genome ― wide transcriptional analysis of the mitotic cell cycle”, Mol. Cell, 2, 1998, pp. 65―73
- [2]P. T. Spellman, G. Sherlock, “Comprehensive identification of cell cycle ― regulated gene of the yeast saccharomyces cerevisiae by microarray hybridation”. Mol. Biol. Cell, 9, 1998, pp. 3273–3297
- [3]S. Raychaudhuri, J. M. Stuart, “Principal components analysis to summarize microarray experiments: application to sporulation time series”, Pac Symp Biocomput 4, 2000, pp. 55–466
- [4]O Alter, P. O. Brown and D. Botstein, “Singular valu decomposition for genome - wide expression data processing and modeling”, PNAS, 97, 2000, pp. 10101–10106
- [5]Y. Yamanishi, M. Itoh and M. Kanehisa, “Extraction of Organism Groups from Phylogenetic Profiles Using Independent Component Analysis”, Genome Informatics13, 2002, pp. 61–70